|
    
~ Pubertal Development Aberrations ~
Delayed or Interrupted Puberty:
ú Definition:
À failure of
development of 2o sex characteristic by 13 y/o, or
Á no menarche by
16 y/o, or
 no attainment of
menarche 5 years or more after onset of puberty
- Anatomic abnormalities of the genital outflow tract
:
- Mullerian dysgenesis (Rokitansdy-Kuster-Hauser
syndrome):
- Distal genital tract obstruction: HSG,
- Imperforate hymen Laparoscopy
- Transverse vaginal septum:
ú Obstruction or
malformation of the distal genital tract must be distinguished
from androgen insensitivity. Individuals with androgen
insensitivity have
breast development in the absence of significant pubic and
axillary hair
development; the vagina may be absent or foreshortened in
these women.
ú Classification
of Mullerian anomalies:
|
Class I: Segmented M ullerian
agenesis or hypoplasia
A. vaginal
B. cervical
C. fundal
D. tubal
E. combined |
|
Class II: Unicornuate uterus
with a rudimentary horn
- with a communicating endometrial cavity
- with a noncommunicating cavity
- with no cavity
- without any rudimentary horn
|
|
Class III: Uterus didelphis |
|
Class IV: Bicornuate uterus
complete to th internal os
partial
arcuate
|
|
Class V: Septate uterus
with a complete septum
with an incomplete septum
|
|
Class VI: Uterus with internal luminal changes |
Hypergonadotropic hypogonadism:
ú FSH >
30 mIU/ml之gonadal “failure”
- Gonadal dysgenesis with stigmata of Turner’s syndrome:
- 45X or mosaic karyotype (45X/45XX or 45X/45XY)
- Turner stigmata (see p783)
- Even in the presence of typical Turner stigmata, a
karyotype is indicated to
eliminate any possibility of any portion of a Y chromosome.
If Y
chromosome is identified, surgical extirpation of the gonad
is warranted to
eliminate any possibility of a germ cell neoplasm.
- Treatment:
? exogenous growth hormone:
dosage: 25% greater than those for growth hormone
deficiency
‚ estrogen:
low dose initially, (eg. 0.3-0.625 conjugated estrogen qd)
at 12-13 y/o
increasing dose within 1-2 years to 2X for those for
menopause
ƒ progestin:
dosage: provera (5) 5-10 mg for 12-14 ds every 1-2 months
- Pure gonadal dysgenesis: streak gonads (nonfunctional gonads)
- 46XX
- 46XY: also called Swyer’s syndrome
- FSH Ý (因streak
gonads不產生steroid及inhibin)
- Treatment:
? surgical extirpation:
for 46XY
‚ exogenous estrogen
therapy
- Early gonadal “failure” with apparent normal ovarian development:
ú Survey: FSH,
prolactin, thyroid funcion, karyotype, etc.
ú 註:
- Gonadotrophin-Releasing Hormone º
GnRH, secreted by hypothalamus
- Gonadotrophin º FSH, LH,
etc., secreted by pituitary gland
- Gonadal Steroid º E2,
etc., secreted by gonadal gland (eg. ovary, testes)
- Hypogonadotropic hypogonadism
:
ú FSH <
10 mIU/ml且LH <
10 mIU/ml之gonadal “failure”
- Constitutional delay:
- the most common cause of delayed puberty
- Isolated gonadotropin deficiency:
- Associated with midline defects (Kallmann’s syndrome):
- Also called olfactogenital dysplasia
- Triade: anosmia, hypogonadism, color blindness
- A disorder of GnRH pulse generator
- X-linked, KALIG-I (Kallmann syndrome Interval Gene I)
- Independent of associated disorders:
- Prader-Labhardt-willi syndrome:
- Laurence-Moon-Bardet-Biedl syndrome:
- Many other rare syndromes:
- Associated with multiple hormone deficiencies:
- GH ß , TSH ß
, usually hypothalamic origin
- Neoplasms of the hypothalamic-pituitary area:
- Craniopharyngiomas
- Pituitary adenomas
- Others
- Infiltrative process (Langerhans-cell type histiocytosis):
- After irradiation of the central nervous system:
- Severe chronic illnesses with malnutrition:
- Anorexia nervosa and related disorders:
- Severe hypothalamic amenorrhea (rare):
- Antidopaminergic and gonadotropin-releasing inhibiting drugs (especially
psychotropic agents, opiates):
- Primary hypothyroidism:
- TRH Ý , è
stimulate prolactin production, è LH ß
and FSH ß
- Cushing’s syndrome
:
- Asynchronous Pubertal Development
:
ú Pubertal
development deviating from the normal pattern
ú the characteristic
of androgen insensitivity (androgen receptor defect)
ú Diagnosis: PE,
normal to elevated testesterone, LH, and FSH
ú Treatment:
gonadectomy, exogenous estrogen
- complete androgen insensitivity syndrome
( testicular feminization):
- Incomplete androgen insensitivity syndrome
:
ú (See figure
23.8, p784)
- Precocious Puberty
:
ú Pubertal
development before 8y/o
ú Isoseual
precocious puberty: changing as the phenotypic sex
Heterosexual precocious puberty: changing as the opposite
sex
ú (See figure
23.16, p794)
- Central (true) precocious puberty
:
- Constitutional (idiopathic) precocious puberty:
- Hypothalamic neoplasms (most commonly hamartomas):
- Congenital malformations:
- Infiltrative process (Langerhans-cell type histiocytosis):
- After irradiation:
- Trauma:
- Infection:
- Precocious puberty of peripheral origin (precocious pseudopuberty)
:
- Gonadotropin-secreting neoplasms:
- Human chorionic gonadotropin-secreting
- Ectopic germinomas (pinealomas)
- Choriocarcinomas
- Teratomas
- Hepatoblastomas
- Luteinizing hormon-secreting (pituitary adenomas)
- Gonadal neoplasms:
- Estrogen-secreting:
- Granulosa-theca cell tumors
- Gonadal sex-cord tumors
- Androgen-secreting:
- Arrhenoblastomas
- Teratomas
- Congenital adrenal hyperplasia:
- 21-Hydroxylase (P450c21) deficiency: the most type
- 11 b -Hydroxylase (P450c11) deficiency
- 3 b -Hydroxysteroid dehydrogenase deficiency
- Treatment:
? hydrocortisone (10-20
mg/m2 body surface area)
‚ mineralocorticoid
replacement
- Adrenal neoplasms:
- Adenomas
- Carcinomas
- Autonomous gonadal hypersecretion:
- Cysts
- McCune-Albright syndrome
- Iatrogenic ingestion/absorption of estrogens or androgens:
- Heterosexual Puberty
:
ú At the expected
age of normal puberty, development as the opposite sex
- Polycystic ovarian syndrome
: the most common cause
- LH-dependent hyperandrogenism
- Nonclassic forms of congenital adrenal hyperplasia
:
- Idiopathic hirsutism
:
- Mixed gonadal dysgenesis
:
- Rare forms of male pseudohermaphroditism (Reifenstein syndrome, 5a
-reductase deficiency)
:
- Cushing’s syndrome (rare)
:
- Androgen-secreting neoplasms (rare)
:
Flow diagram for the evaluation of precocious puberty in
phenotypic females
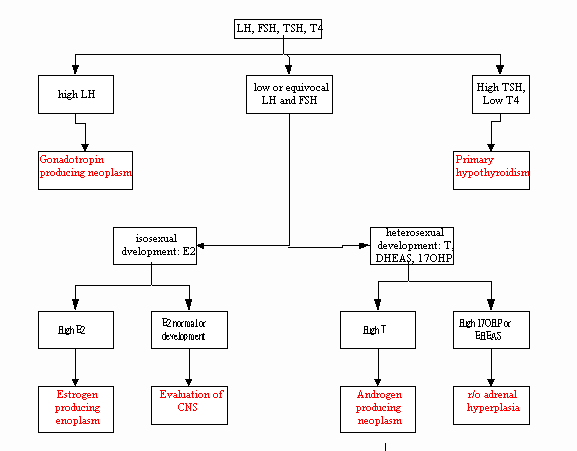
Flow diagram for the evaluation of delayed or interrupted
pubertal development, including primary amenorrhea, in phenotypic females
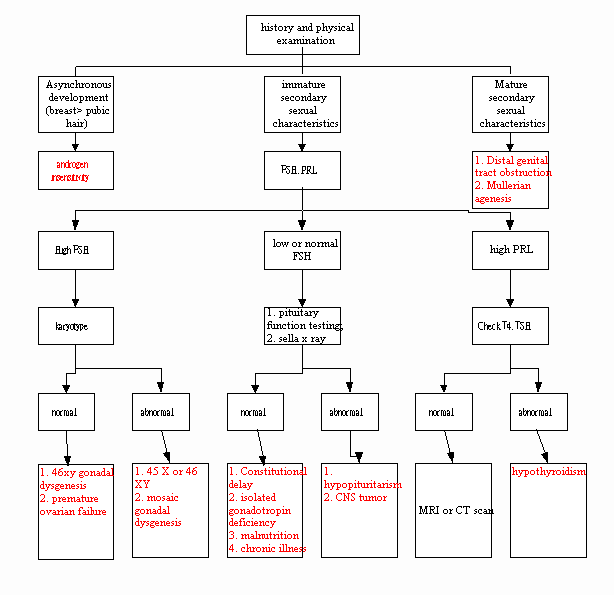
Filename: Pubertal Development Aberrations
| 




